
by Laura Córcoles Gil
There are lots of challenges involved in the study of a crime scene. Forensic scientists are constantly seeking for novel methods that can help in the reconstruction of crime scenes.
Epigenetics is an emerging area of interest in forensic science. With the advances of next-sequencing techniques and the growing DNA methylation datasets together with epigenetic protocols, the prospect of investigating and solving the critical challenges in the study of a crime scene is highly promising, and that’s why new differentially methylated regions are being searched for constantly to serve as markers in forensic DNA ¹².
So, what are DNA methylation applications in the forensic sciences field? Which are the challenges of the use of DNA methylation for forensic analysis? What can we expect in the future?
Introduction
Forensic biology needs to deal with difficulties like discriminating monozygotic twins, predicting age of humans from its left over body tissues and distinguishing body fluids / tissues with challenging identity determination.
That makes forensic scientists seek constantly for novel methods that can help in the reconstruction of crime scenes¹².
Methylation analysis of DNA has proved to have a large range of applications in forensic science such as identifying single or mixed body fluid, discriminating monozygotic twins, age prediction, sex determination, and also phenotypic information of the donor. It has proved to give information about addictions and behavioural prediction as well as race or ethnicity identification. ²
DNA methylation
Epigenetics describes the study of changes in gene expression that occur without alteration to the DNA sequence. In humans, the most widely studied epigenetic modification is DNA methylation and usually occurs in CpG nucleotides¹³.
This epigenetic mark is really important for normal development in the human
genome. The loss of DNA methylation leads to apoptosis or growth arrest in
normal cells. It is generally believed that methylated DNA is associated with condensed chromatin structure and with that repression of gene transcription but there are also evidences where DNA methylation is associated with gene activation or where no relationship between methylation status and gene expression has been found. This evidences the high degree of complexity involved in determining the phenotypic effects of DNA methylation ¹ ¹³.
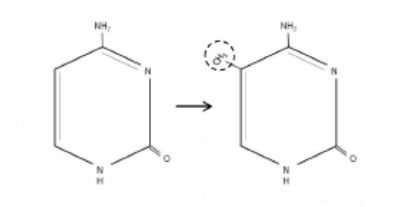
This methylation involves the addition of a methyl group at the 5’ position of cytosine residues (Figure 1) ¹ ² ³ ¹³.
Figure 1. DNA methylation
CpG islands generally act as strong promoters and are also thought to serve as replication origins. DNA methyltransferases are enzymes responsible for de novo methylation and maintenance of methylation. 1,2. (Adapted from Vidaki et al. 2013).
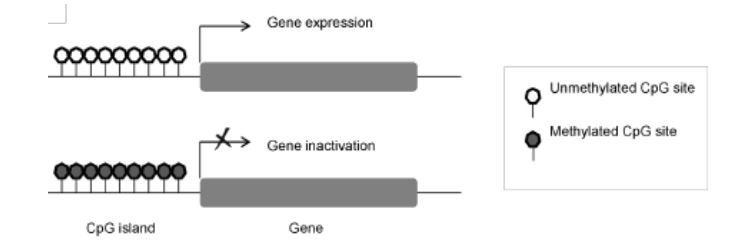
DNA methylation and gene expression
A key role of DNA methylation is to control gene expression. It’s been seen that there is a correlation between gene expression and DNA methylation of CpG promoter regions. Aberration in DNA methylation has long been linked to diseases such as cancer. ¹²
Figure 2. Schematic representation of a CpG island
Adapted from Vidaki et al. 2013
Environmental influences in dna methylation
It is considered a stable epigenetic modification, because unlike protein or RNA markers, which are quickly degraded, DNA methylation is not thus allowing for quantitative analysis of older samples. Although it’s stability, DNA methylation is influenced by various factors such as nutrition, early life experiences, ageing, exposure to pollutants as well as social environments. ¹ ².
With the ages, DNA methylation decreases due to the loss of the ability of the DNA methylation machinery to maintain methylation patterns.
Various food components have been observed to alter gene expression by changing DNA methylation. Tobacco smoking, psychosocial factors such as cortisol output and perceived stress, for example, have been related with DNA methylation patterns. Exposure to chemical pollutants of the environment also induces alterations in DNA methylation. ¹.
Differential DNA methylation
In a crime scene, the body fluids of a human such as blood, semen, visceral fluids, vaginal fluids and saliva for example, are washed out of its natural texture and are often mixed or discoloured due to drying/bleaching out during a long exposure. These results in a challenge to determine the actual cause/severity of the crime. ²
Genome-wide methylation analysis has discovered numerous regions with different levels of methylation in various cell types and tissues.
DNA methylation profiles are tissue-specific. That means that every tissue or cell type shows differentially methylation patterns. This fact makes this methylation profiles useful for differentiating which type of tissue do we have as an evidence of a crime ¹ ².
The analysis of these differentially methylated regions represents a new and reliable technique to identify the fluids and tissues found at the scene of a violent act.²
Application of DNA methylation in forensic sciences
Of all the numerous chromatin marks that comprise the epigenome, DNA methylation is the most accessible and characterised. It is also an extremely stable biological macromolecule. All these characteristics make the DNA methylation status of a gene an attractive detection and diagnostic biomarker that serves as a perfect target for studies. ¹
Although CpG methylation is chemically stable, studies have indicated that the pattern and amount of methylation can change over the lifetime. This occurs in response to dietary and lifestyle factors such as B vitamin, alcohol and tobacco consumption and physical exercise, and biological factors such as age. That evidences that DNA methylation differences are an interface between the dynamic environment and the fixed genome ¹³.
The methods for analysing the DNA methylation target only extracted DNA, so there is a minimal consumption or destruction of physical material, and that is essential in criminal cases. At the same time, with a single assay it is possible to analyse multiple tissues ¹.
With sodium bisulphite treatment (Figure 3) that converts unmethylated cytosines to uracils but doesn’t affect methylated cytosines, methylated and unmethylated cytosines can be discriminated. With the subsequent PCR 3 amplification, cloning and Sanger sequencing, the methylation status of the loci can be determined ² ³.
Methylation patterns are used to determine if a trace of DNA located in a crime scene has credibility or not. And that is extremely important in forensic investigations¹.
Figure 3
Bisulphite conversion of non-methylated and methylated DNA sequences. Only non- methylated cytosines are converted into uracil. The methylated remain unchanged. Adapted from Vidaki et al.
Tissue sources and body fluid identification
In forensic sciences, detection and identification of a body fluid present at a crime scene is crucial for the investigation. DNA methylation is cell-type specific and that means that each cell-type has a DNA methylation profile. That allows not only to find the origin of the DNA sample, but also to quantify the population of various cell types in the sample. 1,2,6,9,13.
Frumkin et al, where the first to highlight in 2011 the use of differential CpG methylation for semen identification in mixtures. More studies have been made until that moment to describe various epigenetic markers and analysis
methods for different tissues like blood, saliva, semen, vaginal material and menstrual fluid. That possibility to discriminate all this tissues from DNA obtained from a mixture of bodily fluid is called Next generation serology. 2,9,13.
Candidate CpG sites need to display minimal inter and intraindividual variation in methylation levels for the same tissue type and also have significantly different methylation between the target tissue and all other forensically relevant body fluids under different environmental conditions because if not, they loose all the value.
Age prediction
Determining the age of victims and suspects is important in forensic investigations. Scientists have identified genome regions whose DNA
methylation pattern is age-sensitive. Using these loci, biological age could be predicted as precise as within 5 years of chronological age. 1,2,4,5,10.
CpG sites associated with age have been detected in genes such as ELOVL2, C2orf132, TRIM59, FHL2, ASPA, KLF14, PDE4C, EDARDD, NOX4 and TTC7B. Methylation markers associated with the ELOVL2 gene in multiple studies in different populations cite high correlations between methylation level
and age. 13.
DNA methylation can also be used to determine aging rate. This has to take in account that there could be indicators of underlying health or physical conditions that may interfere the conclusions. 1,4,5,10.
With the ages, DNA methylation machinery loses its ability to maintain
methylation patterns across cellular divisions. That means that with age, there is a general decrease in DNA methylation with hypermethylation of certain gene promoters within the genome. 1,2.
Differentiation of monozygotic twins
Twins present a challenge, because despite various phenotypic differences, identical twins carry the same DNA because they develop from a single fertilized egg.
It’s been found that DNA methylation has some variation between monozygotic twins. This variation is due to environmental differences.
This DNA methylation differences provide an alternative for positive identifications. 1,2,7,8, 11
The development of standard detection methods is underway. Studies have evaluated the use of LINE1 elements. This highly repeated sequences methylation levels are representative of the methylation level of the whole 5 genome. LINE1 has proved to differentiate between 12.61% of monozygotic pairs, and that confirms that can be used to differentiate between monozygotic twins, but other methylation markers need to be found with higher discriminating percentage. 11
These differences at an epigenetic level may help to know which twin is the trace donor but we still need to compare the trace with the two twins. It is expected that additional research testing the stability of DNA methylation differences will determine if DNA methylation can solve this question in the future.12
The number and magnitude of epigenetic differences needed to achieve a valid identification remains unclear. There are evidences that indicate that this could depend on age and/or environment. More research needs to be done to investigate if differential DNA methylation is suitable for monozygotic twin discrimination.13
Biogeographic / Ethnic Differences
There are differences in DNA methylation patterns between different human populations right from the birth. These differences persist for generations as long as the person stays in the same biogeographic region.
Methylation studies have shown distinct pattern of CpG methylation at certain locus in the autosomal DNA between different human populations or races.
It’s been seen that the main factors influencing spatial epigenetic variation are nutrients, UVA exposure and pathogens. Besides geographic ancestry information from genetic markers, it is thought that epigenetic profiling is going to give additional residence information in the near future.12
DNA methylation challenges
DNA methylation analysis is a really promising method for solving some of the challenges in solving crimes, but it also has its challenges. 6
To be accepted as court evidence, the sensitivity of the marker used and the detection methods need to be fully considered. 1.
Forensic DNA analysis has requirements related with quality and quantity. In a crime scene, traces are usually of low quality and quantity, and that is a problem. In epigenetic analyses this is also a problem, because it has consequences for the type and number of markers that can be analysed, and the technology that can be used. 12
One single marker does not deliver enough forensically useful information so we need to use multiplex genotyping methods to analyse several epigenetic markers at once. However, currently available technologies for this simultaneous analysis such as DNA methylation microarrays and whole- genome bisulfite sequencing, are not suitable due to the large input amounts of high-quality DNA they require.
Current epigenetic analysis technologies able to deal with low-quality / quantity DNA, such as bisulfite pyrosequencing, methylation quantitative PCR, and
EPITYPER, are limited in their multiplexing capacities (fewer than 20 markers), and this is usually insufficient to address a forensic question of interest.12
Due to the low amounts of DNA obtained from a crime scene, highly sensitive technologies are needed for reliable detection of DNA methylation variation.
Methods like methylation SNaPshot with multiplexing capacity currently have sensitivities down to a few nanograms of DNA input per PCR but most current epigenetic methodologies require bisulfite conversion prior to marker analysis, and this bisulfite conversion typically requires minimum 50-200 ng for reliable performance. Highly sensitive technologies allowing simultaneous analysis of large numbers of DNA methylation markers from low-quantity/quality DNA are still not known.12
It is also important to evaluate which method of analysis, amplification and
design of assay are we going to use, because each method will present its own rewards and limitations.1 7
Tests using epigenetic markers need to be standardised with high sensitivity before they are admissible in court.12
DNA methylation assays have helped to unequivocally identify various body fluids. But, there are factors that have to be considered before. It is really important to examine several samples to certify that the CpG site we are selecting does not have low levels of inter-individual variation.
It is also important to be sure that the CpG site is not influenced by external stimuli or is age-dependent.1,13.
It’s been proved that DNA methylation machinery loses its ability to maintain methylation patterns across cellular division, so with ages, DNA methylation decreases. That makes this markers useful for age determination, but it has to be taken in account that there could be indicators of underlying health or physical conditions that may interfere the conclusions and give as the wrong age. 1,4,5,10.
It’s been seen that Monozygotic twins have methylation differences due to environmental differences, and that could help forensics know which twin is the trace donor. But the number and magnitude of epigenetic differences needed to achieve a valid identification remains unclear. More research needs to be done to see if DNA methylation patterns can solve this monozygotic twin problem.13
So, we would say that the use of DNA methylation in forensic investigations is still risky considering that the amount of genomic DNA obtained is sometimes challenging, the sensitivity of the analysis method is not enough, and the selected marker sensitivity is also important.1, 2.
Conclusions
The epigenetic field has appeared in the forensic sciences with high impact promising to solve some of the challenges that forensic biology is dealing with.
DNA methylation, that without altering the DNA sequence leads to phenotypic variation among tissues, monozygotic twins and humans, provides a new 8 method to distinguish cell types or tissues, the age of humans and identical twin differentiation. All this information could be used to complement the currently existing forensic technologies accepted as court evidence but there is not yet an agreed methodology for targeted detection and analysis of DNA methylation markers in forensic research.
Tests based on methylation variation have been proposed for age estimation of DNA donor, monozygotic twins and body fluid identification, and it is hypothesized that in the future it is also going to be useful for lifestyle predictions and biogeographic ancestry determination.
As the best CpG sites for determining DNA methylation levels are discovered, the forensic methodologies will become easier to put into practice.
The future of DNA methylation analysis in forensics will surely be constantly evolving and will most likely be poles apart from what we see today.
In the future, and with more research in this topic, from a drop of blood at a crime scene, Scientifics will be able to create a complete profile of the owner of that drop, obtaining details of the age, smoking status, drugs consumed, polluted environment in the living area and even traumatic history of the person.
Forensic DNA methylation profiling has the potential to add value in criminal investigations but there is still more investigation to be done.
References
- Kader F., Ghai M. (2015). DNA methylation and application in forensic sciences. Forensic
Science International, 249:255-265.
- Rana A.K. (2018). Crime investigation through DNA methylation analysis: methods and
applications in forensics. Egyptian journal of forensic sciences. 8:7.
- Frumkin, D., Wasserstrom, A., Davidson, A., and Grafit, A. (2010). Authentication of forensic
DNA samples. Forensic science international Genetics 4, 95-103.
- Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., Klotzle, B., Bibikova,
M., Fan, J. B., Gao, Y., et al. (2013). Genome-wide methylation profiles reveal quantitative
views of human aging rates. Molecular cell 49, 359-367.
9
- Weidner, C. I., Lin, Q., Koch, C. M., Eisele, L., Beier, F., Ziegler, P., Bauerschlag, D. O.,
Jockel, K. H., Erbel, R., Muhleisen, T. W., et al. (2014). Aging of blood can be tracked by DNA
methylation changes at just three CpG sites. Genome biology 15, R24.
- Vidaki, A., Daniel, B., and Court, D. S. (2013). Forensic DNA methylation profiling–potential
opportunities and challenges. Forensic science international Genetics 7, 499-507.
- Gordon, L., Joo, J. E., Powell, J. E., Ollikainen, M., Novakovic, B., Li, X., Andronikos, R.,
Cruickshank, M. N., Conneely, K. N., Smith, A. K., et al. (2012). Neonatal DNA methylation
profile in human twins is specified by a complex interplay between intrauterine environmental
and genetic factors, subject to tissue-specific influence. Genome research 22, 1395-1406.
- Levesque, M. L., Casey, K. F., Szyf, M., Ismaylova, E., Ly, V., Verner, M. P., Suderman, M.,
Brendgen, M., Vitaro, F., Dionne, G., et al. (2014). Genome-wide DNA methylation variability in
adolescent monozygotic twins followed since birth. Epigenetics : official journal of the DNA
Methylation Society 9, 1410-1421.
- Silva, D. et al. (2016). Developmental validation studies of epigenetic DNA methylation
markers for the detection of blood, semen and saliva samples. Forensic Science International,
23, pp. 55-63.
- Vidaki, A. et al. (2017). DNA Methylation-based forensic age prediction using artificial neural
networks and next generation sequencing. Forensic Science International, 28, pp. 225-236.
- Xu, J. et al. (2015). LINE-1 DNA methylation: A potential forensic marker for discriminating
monozygotic twins. Forensic Science International, 19, pp. 136-145.
- Vidaki A., Kayser M. (2017). From forensic epigenetics to forensic epigenomics: broadening
DNA investigative intelligence. Vidaki and Kayser Genome Biology, 18:238.
- Richards R., Patel J., Stevenson K., Harbison S. (2018). Evaluation of massively parallel
sequencing for forensic DNA methylation profiling. Electrophoresis journal, 0,1-8.

{kind=link}